Medizinprodukte-Hersteller sind verpflichtet, klinische Daten vor und nach der Zulassung ihrer Produkte systematisch zu sammeln und auszuwerten.
Die EU-Medizinprodukteverordnung MDR hat die Anforderungen an den Umfang und die Qualität dieser klinischen Daten erhöht.
Dieser Artikel verschafft Ihnen einen Überblick über die regulatorischen Anforderungen und gibt Tipps, welche klinischen Daten Medizinproduktehersteller auf welche Weise sammeln und bewerten können, um regulatorische Probleme zu vermeiden. Er erwähnt auch, wann auf klinische Daten verzichtet werden kann.
Viele Hersteller sind sich nicht bewusst, welche Bedeutung die klinischen Daten für den Nachweis der Konformität ihrer Produkte haben. Das betrifft sowohl die Zulassung der Produkte als auch die Post-Market-Aktivitäten, insbesondere das „Post-Market Clinical Follow-up“.
1. Klinische Daten: Grundlagen
1.1 Definition
Ähnlich wie die MEDDEV 2.7/1 definiert die Medizinprodukteverordnung MDR den Begriff „klinische Daten“ wie folgt:
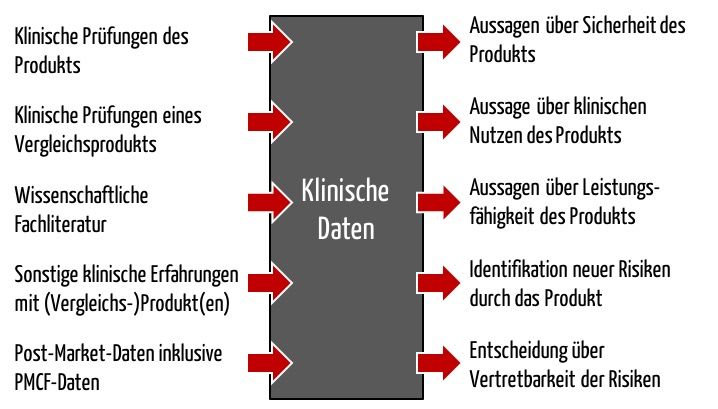
„Angaben zur Sicherheit oder Leistung, die im Rahmen der Anwendung eines Produkts gewonnen werden und die aus den folgenden Quellen stammen:
- klinische Prüfung(en) des betreffenden Produkts,
- klinische Prüfung(en) oder sonstige in der wissenschaftlichen Fachliteratur wiedergegebene Studien über ein Produkt, dessen Gleichartigkeit mit dem betreffenden Produkt nachgewiesen werden kann,
- in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichte Berichte über sonstige klinische Erfahrungen entweder mit dem betreffenden Produkt oder einem Produkt, dessen Gleichartigkeit mit dem betreffenden Produkt nachgewiesen werden kann,
- klinisch relevante Angaben aus der Überwachung nach dem Inverkehrbringen, insbesondere aus der klinischen Nachbeobachtung nach dem Inverkehrbringen;“
Abschnitt 48 in Artikel 2 MDR
1.2 Zwecke, die mit klinischen Daten erreicht werden sollen
Diese Definition nennt sowohl den Zweck als auch die Quellen medizinischer Daten.
Der Anhang VI offenbart, dass der Begriff „Leistung“ im doppelten Sinn zu verstehen ist:
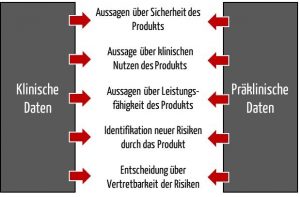
- Die Leistung im Sinne einer Leistungsfähigkeit in Form spezifizierter Leistungsangaben. Ein Beispiel ist die Fähigkeit eines Defibrillators, eine gewisse Energiemenge pro Zeiteinheit abzugeben.
- Die Leistung im Sinne des klinischen Nutzens und des Erreichens der Zweckbestimmung des Produkts. Beim Defibrillator wäre dies, das Herzkammerflimmern zu beenden.
Den Nachweis, dass die spezifizierten Leistungsangaben (1. Punkt) erfüllt sind, erbringen Hersteller meist anhand vorklinischer Daten, die auch präklinische Daten genannt werden. Beispiele für präklinische Daten finden Sie weiter unten.
Im Anhang XIV offenbart die MDR weitere Zwecke klinischer Daten:
- Sie sollen dazu dienen, bisher nicht erkannte Risiken oder Gefährdungen zu identifizieren und neu zu bewerten.
- Anhand aktualisierter klinischer Daten sollen die Hersteller ihre Entscheidung über die Vertretbarkeit der Risiken neu treffen.
Sie stehen noch am Anfang Ihrer klinischen Bewertung?
Unser Clinical Evaluation Jumpstart Kit hilft Ihnen, die ersten Schritte innerhalb der klinischen Bewertung Ihrer Medizinprodukte zu gehen und verschafft Orientierung, was konkret von Ihnen verlangt wird.
Jetzt kostenlos anfordern
2. Quellen von Daten
Für die klinische Bewertung müssen Hersteller nicht nur die klinischen, sondern auch die vorklinischen (präklinischen) Daten berücksichtigen.
2.1 Quellen für klinische Daten
| Daten | Beispiele |
| Klinische Prüfungen zum Produkt |
|
| Klinische Prüfungen eines Äquivalenzprodukts |
|
| Wissenschaftliche Fachliteratur | |
| Sonstige klinische Erfahrungen mit (Äquivalenz-)Produkten |
|
| Post-Market-Daten inklusive PMCF-Daten (Klinische Nachbeobachtung) |
|
2.2 Quellen für präklinische Daten
Zu den Beispielen für präklinische Daten zählen die Ergebnisse folgender Prüfungen:
Daten aus z. B. Usability-Tests, die nach der Inverkehrbringung gesammelt werden und die klinisch relevante Informationen enthalten, zählen definitionsgemäß ebenfalls zu den klinischen Daten.
3. Regulatorische Anforderungen an klinische Daten
3.1 Europa
Die Medizinprodukterichtlinie MDD fordert ebenso wie die Medizinprodukteverordnung MDR, dass die Hersteller klinische Daten sammeln und bewerten. Bei der MDR finden sich diese Forderungen u. a. in den folgenden Kapiteln:
- Artikel 61 fordert eine klinische Bewertung, die sich auf „ausreichende klinische Daten stützt“. Darin heißt es weiter: „Die klinische Bewertung und die dazugehörigen Unterlagen sind während des gesamten Lebenszyklus des Produkts anhand der klinischen Daten zu aktualisieren.“ Die Frequenz hängt von der Klasse des Produkts ab und ist in Artikel 84 der MDR spezifiziert.
- Anhang II (Technische Dokumentation) fordert in Kapitel 6 (Verifizierung und Validierung des Produkts) vorklinische und klinische Daten.
- Besonders ausführlich geht der Anhang XIV Teil A (klinische Bewertung) auf die klinischen Daten ein.
- Anhang XIV Teil B (klinische Nachbeobachtung, PMCF) verlangt von den Herstellern, „auf proaktive Weise klinische Daten“ zu sammeln und zu bewerten.
Die MDR verlangt von den Herstellern, klinische Daten vor und nach der Zulassung zu sammeln. Sie gibt bereits in der Definition vor, welche Quellen die Hersteller mindestens zu berücksichtigen haben.
Wie wichtig der Medizinprodukteverordnung die klinischen Daten sind, offenbart der Anhang VII, der die Anforderungen an die Benannten Stellen spezifiziert:
„Die Benannte Stelle […] schenkt den klinischen Daten aus der Überwachung nach dem Inverkehrbringen und [… der] klinischen Nachbeobachtung […] besondere Aufmerksamkeit […]“.
3.1.1 Klinische Daten von anderen Medizinprodukten
Hersteller dürfen sich laut Anhang XIV bei der klinischen Bewertung nur dann auf klinische Daten anderer Produkte stützen, wenn die Gleichartigkeit dieser anderen Produkte nachgewiesen wird. Die MDR fordert eine technische, biologische und klinische Äquivalenz. Mehr dazu lesen Sie weiter unten.
3.1.2 Verzicht auf klinische Daten
MDR und MDD erlauben lediglich unter ganz bestimmten Voraussetzungen, in der klinischen Bewertung auf klinische Daten zu verzichten.
In der MDD gab es in Anhang X, Absatz 1.1d bereits eine Ausnahmeregelung für Produkte, bei denen die Bewertung auf Basis klinischer Daten „für nicht notwendig erachtet“ wird.
Die MDR erlaubt diese Ausnahme, wenn der Nachweis mittels klinischer Daten als ungeeignet oder als nicht angemessen erachtet wird („is not deemed appropriate“).
Wird der Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen auf der Grundlage klinischer Daten für ungeeignet erachtet, ist jede solche Ausnahme auf der Grundlage des Risikomanagements des Herstellers und unter Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper, der bezweckten klinischen Leistung und der Angaben des Herstellers angemessen zu begründen; dies gilt unbeschadet des Absatzes 4. In diesem Fall muss der Hersteller in der technischen Dokumentation gemäß Anhang II gebührend begründen, warum er den Nachweis der Übereinstimmung mit grundlegenden Sicherheits- und Leistungsanforderungen allein auf der Grundlage der Ergebnisse nichtklinischer Testmethoden, einschließlich Leistungsbewertung, technischer Prüfung („bench testing“) und vorklinischer Bewertung, für geeignet hält.
MDR, Artikel 61, Satz (10)
Hilfsmittel (z. B. Mundspatel) oder Werkzeuge (z. B. Skalpell, Zahnarztbohrer) können oft ohne produktspezifische klinische Daten bewertet werden, da eine solitäre klinische Prüfung nicht angemessen oder ethisch nicht vertretbar erscheint. Natürlich muss dies nachvollziehbar begründet werden.
Ein Verzicht auf klinische Daten muss nachvollziehbar begründet werden. Für die Begründung können Sie z. B. über allgemeine klinische Daten im State of the Art nachweisen, dass Ihr Gerät auf einer bewährten Technologie basiert, die dem anerkannten Stand der Technik entspricht und allgemein wenig Änderung erfährt. Idealerweise liegen Produktnormen vor, in denen Leistungsparameter angegeben werden, durch deren Einhaltung die Sicherheit gewährleistet werden kann. Der Nutzen kann z. B. über Leitlinien medizinischer Fachgesellschaften nachgewiesen werden.
Die MDCG beschreibt diese Möglichkeit nur im Dokument MDCG 2020-6 für Bestandsprodukte (englisch: legacy devices). Wir denken aber, dass der Grundgedanke auch anwendbar ist, wenn Sie ein derartiges Produkt neu in Ihr Portfolio aufnehmen und erstmals zulassen.
Noch präziser beschreibt das TeamNB in seinem Positionspapier „Clinical Evaluation Based on Non-Clinical Data: EU Medical Device Regulation 2017/745 Article 61.10“ die Voraussetzung für die Ausnahmen.
Demnach definiert der entsprechende Artikel 61(10) einen Ausnahmeweg, der nur die Datenart ändert, nicht aber die Ziele der klinischen Bewertung. Das Papier liefert detaillierte Fragenkataloge zu den drei Bewertungsbereichen klinische Leistung/Claims, Körperinteraktion und Risikomanagement, anhand derer Benannte Stellen die Begründung des Herstellers prüfen werden.
Die Begründung muss so detailliert sein, dass auch Unbeteiligte nachvollziehen können, warum klinische Daten nicht geeignet sind – wobei Argumente wie „Daten nicht verfügbar“ oder „schwer zu generieren“ ausdrücklich nicht akzeptiert werden.
Hersteller sollten daher jetzt ihre State-of-the-Art-Analysen, Artikel-61(10)-Begründungen, die Definition klinischer Benefits (auch indirekte) sowie ihre PMS/PMCF-Strategien systematisch im Hinblick auf die neuen Anforderungen prüfen und gegebenenfalls in einen strukturierten Dialog mit ihrer Benannten Stelle treten.
Beachten Sie, dass ein Verzicht auf klinische Daten nicht bedeutet, dass Sie Ihr Produkt nicht schon vor der Inverkehrbringung einer Prüfung durch Anwender unterziehen müssen. In den meisten Fällen brauchen Sie summative Tests, eine sogenannte Usability-Studie, um die Gebrauchstauglichkeit zu validieren.
3.2 USA / FDA
Die FDA verlangt von den Herstellern bei den Zulassungen, z. B. den Premarket Notifications (PMN) oder Premarket Approvals (PMA), klinische Daten. Bei den PMN (510(k)) stammen diese in der Regel vom Vergleichsprodukt, wie die FDA selbst sagt.
Ende Februar 2018 hat die FDA ein Guidance-Dokument veröffentlicht, das beschreibt, in welcher Form die FDA welche klinischen Daten akzeptiert. Es trägt den Titel Acceptance of Clinical Data to Support Medical Device Applications and Submissions Frequently Asked Questions.
2023 hat die FDA das Guidance-Dokument Recommendations for the Use of Clinical Data in Premarket Notification [510(k)] Submissions veröffentlicht.
4. Typische Probleme
4.1 Äquivalenz der Produkte
Die meisten Medizinproduktehersteller sind bestrebt, klinische Prüfungen mit dem eigenen Produkt zu vermeiden. Klinische Studien sind teuer und benötigen meistens Monate. Daher versuchen die Hersteller, die klinischen Daten von Vergleichsprodukten zu verwenden und/oder den Beweis nach dem „Literaturverfahren“ zu führen.
Allerdings müssen diese Vergleichsprodukte „ausreichend ähnlich“ sein. Die MDR und die MEDDEV 2.7/1 Revision 4 haben die Anforderungen an diese Äquivalenz deutlich erhöht. Sie nennen drei Aspekte:
- Technisch: Das Produkt ist von ähnlicher Bauart, wird unter ähnlichen Anwendungsbedingungen angewandt, hat ähnliche Spezifikationen und Eigenschaften einschließlich physikalisch-chemischer Eigenschaften wie Energieintensität, Zugfestigkeit, Viskosität, Oberflächenbeschaffenheit, Wellenlänge und Software-Algorithmen, verwendet gegebenenfalls ähnliche Entwicklungsmethoden und hat ähnliche Funktionsgrundsätze und entscheidende Leistungsanforderungen.
- Biologisch: Das Produkt verwendet die gleichen Materialien oder Stoffe im Kontakt mit den gleichen menschlichen Geweben oder Körperflüssigkeiten für eine ähnliche Art und Dauer des Kontakts bei ähnlichem Abgabeverhalten der Stoffe einschließlich Abbauprodukte und herauslösbarer Bestandteile („leachables“).
- Klinisch: Das Produkt wird unter der gleichen klinischen Bedingung oder zum gleichen klinischen Zweck, einschließlich eines ähnlichen Schweregrads und Stadiums der Krankheit, an der gleichen Körperstelle und bei ähnlichen Patientenpopulationen in Bezug auf u.a. Alter, Anatomie und Physiologie angewandt, hat die gleichen Anwender und erbringt eine ähnliche, maßgebliche und entscheidende Leistung im Hinblick auf die erwartete klinische Wirkung für eine spezielle Zweckbestimmung. (Quelle MDR)
Das Johner Institut erlebt regelmäßig, dass Benannte Stellen die Anforderungen an die Äquivalenz so hoch auslegen, dass klinische Prüfungen unumgänglich werden. Das ist besonders bei unkritischen Produkten nicht immer nachvollziehbar und sinnvoll.
4.2 Quantität und Qualität der Daten
Die Daten müssen „in quantitativer und qualitativer Hinsicht ausreichend“ sein, um „qualifiziert beurteilen zu können, ob das Produkt sicher ist und den angestrebten klinischen Nutzen […] erreicht“. Dazu müssen die klinischen Daten „wissenschaftlich fundiert, zuverlässig und solide“ sein.
Die Prüfer der Benannten Stellen legen auf folgende Eigenschaften besonderen Wert:
- Die klinischen Daten müssen quantitativ ausreichend sein, um eine statistische Signifikanz zu belegen.
- Sie müssen aus Quellen stammen, deren wissenschaftliche Validität keinen Zweifel aufwirft.
- Die Daten verschiedener Quellen sollten die These des Herstellers belegen und damit den Nachweis des Nutzens, der Leistung und der Sicherheit des Produkts erbringen.
- Es müssen möglichst alle Quellen durchsucht bzw. ausgewertet werden. Manche Hersteller wehren sich gegen die Forderung, die kostenpflichtige Datenbank EMBASE zu nutzen. Es gibt keine Anforderung, alle Datenquellen zu verwenden. PubMed ist von den meisten benannten Stellen als alleinige Quelle anerkannt.
- Die Daten müssen mit Produkten, Technologien oder Verfahren gewonnen werden, bei denen die Anforderungen an die Äquivalenz und damit Vergleichbarkeit erfüllt sind.
Besonders die Beurteilung der wissenschaftlichen Validität klinischer Daten führt zu Diskussionen zwischen Herstellern einerseits und Benannten Stellen und Behörden andererseits. Dabei geht es um solche Fragen:
- Ist das wissenschaftliche Journal seriös? Der Impact Factor des Journals ist dabei eine relevante Metrik.
- Ist das Studiendesign angemessen? Müssen die Daten aus einer prospektiven, randomisierten und kontrollierten Studie stammen?
- Hat die Studie einen Bias? Hat die Auswahl der Probanden oder des Verfahrens oder sogar der Hersteller selbst einen Einfluss auf die Ergebnisse (genommen)?
Diese wissenschaftliche Güte von Publikationen versucht der GRADE, ein „Level of Evidence“ nach Cochrane, zu quantifizieren.
4.3 Verpflichtung zu klinischen Prüfungen
Insbesondere bei Implantaten der Klasse IIb und allen Produkten der Klasse III besteht die MDR darauf, dass die Hersteller die klinischen Daten (auch) im Rahmen klinischer Prüfungen mit dem speziellen Produkt erheben. Ausnahmeregelungen existieren zwar, aber die Anzahl klinischer Prüfungen wird zunehmen.
5. Tipps und Fazit
Die Anforderungen an die Hersteller steigen, den Nutzen und die Sicherheit ihrer Produkte anhand klinischer Daten wissenschaftlich zu beweisen. Das Sammeln der wissenschaftlichen Daten, z. B. in Form einer klinischen Prüfung, darf aber nicht zum Selbstzweck werden. Wir empfehlen daher:
5.1 Auf Leistungsdaten ausweichen (falls möglich)
Die EU-Richtlinien bzw. Verordnungen erlauben es (MDD Anhang X, Absatz 1.1d bzw. Kapitel VI, Artikel 61 Abs. 10 der MDR), in gewissen Fällen statt klinischer Daten auch präklinische Daten wie Leistungsdaten, Simulationsergebnisse und Systemtests zu verwenden. Dies sollte besonders bei unkritischen Produkten ohne (oder mit nur wenig) Rückwirkung mit dem Patienten gelingen. Beispiele für solche Produkte, bei denen es dem Team des Johner Instituts gelang, diesen Weg zu gehen, sind: Stand-alone-Software, Mundspatel, Behandlungsstühle, sehr etablierte stoffliche Medizinprodukte und sehr etablierte kardiologische Elektroden.
5.2 Datenbank klinischer Prüfungen beachten
Die wissenschaftliche Fachliteratur müssen Hersteller in jedem Fall beleuchten. Wir empfehlen zudem, Registries und die Datenbanken für klinische Prüfungen wie zu berücksichtigen.
5.3 Daten von Vorgängerprodukten nutzen
Viele Firmen unterschätzen den Wert der Daten, die von Vorgängerprodukten stammen. Manchmal ist es einfacher, eine nicht-interventionelle Anwendungsstudie mit solch einem Vorgängerprodukt durchzuführen, als eine neue klinische Prüfung aufzusetzen. Stimmen Sie mit Ihrer Benannten Stelle diesen Ansatz ab.
5.4 Sofort mit dem Sammeln der Daten beginnen
Mit der MDR müssen Sie Ihre Produkte neu zulassen. Nutzen Sie die Zeit bis dahin, um mit den bereits zugelassenen Produkten ausreichend Daten zu sammeln, auf die Sie später zurückgreifen und damit eine klinische Prüfung vermeiden können.
Die Zweckbestimmung stellt eine Voraussetzung für jede klinische Bewertung dar. Diese Zweckbestimmung sollte möglichst quantitativ den beabsichtigten klinischen Nutzen spezifizieren.
Kontaktieren Sie gerne das Johner Institut, wenn Ihre Benannte Stelle eine zeit- und kostenintensive klinische Prüfung verlangt, obwohl Sie der Meinung sind, über ausreichend klinische Daten zu verfügen. Regelmäßig finden unsere klinischen Experten einen Ausweg. Sie helfen Ihnen auch, eine klinische Prüfung oder Anwendungsbeobachtung schnell, planbar, kosteneffizient und gesetzeskonform durchzuführen.
Kontakt aufnehmen
Änderungshistorie
- 2026-05-07: Positionspapier des TeamNB aufgegriffen
- Satz im dritter Abschnitt der Einleitung ergänzt
- Vier Abschnitte im Kapitel 3.1.2 (oberhalb des Kastens) ergänzt
- 2024-04-17: Kapitel 3.1.2 eingefügt und Nummerierung geändert
- 2023-10-23: Artikel aktualisiert; z. B. durch Hinweis auf neues FDA Guidance Document. Kapitel 1 und 5 neu strukturiert.
- 2018-09-12: Erste Version veröffentlicht
Ähnliche Beiträge
PakarPBN
A Private Blog Network (PBN) is a collection of websites that are controlled by a single individual or organization and used primarily to build backlinks to a “money site” in order to influence its ranking in search engines such as Google. The core idea behind a PBN is based on the importance of backlinks in Google’s ranking algorithm. Since Google views backlinks as signals of authority and trust, some website owners attempt to artificially create these signals through a controlled network of sites.
In a typical PBN setup, the owner acquires expired or aged domains that already have existing authority, backlinks, and history. These domains are rebuilt with new content and hosted separately, often using different IP addresses, hosting providers, themes, and ownership details to make them appear unrelated. Within the content published on these sites, links are strategically placed that point to the main website the owner wants to rank higher. By doing this, the owner attempts to pass link equity (also known as “link juice”) from the PBN sites to the target website.
The purpose of a PBN is to give the impression that the target website is naturally earning links from multiple independent sources. If done effectively, this can temporarily improve keyword rankings, increase organic visibility, and drive more traffic from search results.