Die Medizinprodukteverordnung hat die Anforderungen an die Händler deutlich erhöht. Lernen Sie diese Anforderungen zu verstehen, um die mehrjährigen Freiheitsstrafen zu vermeiden, die bei einem Verstoß drohen.
Dieser Beitrag berücksichtigt auch eine umfangreiche Leitlinie der irischen Aufsichtsbehörde.
1. Händler: Definition & Abgrenzung
a) Definition
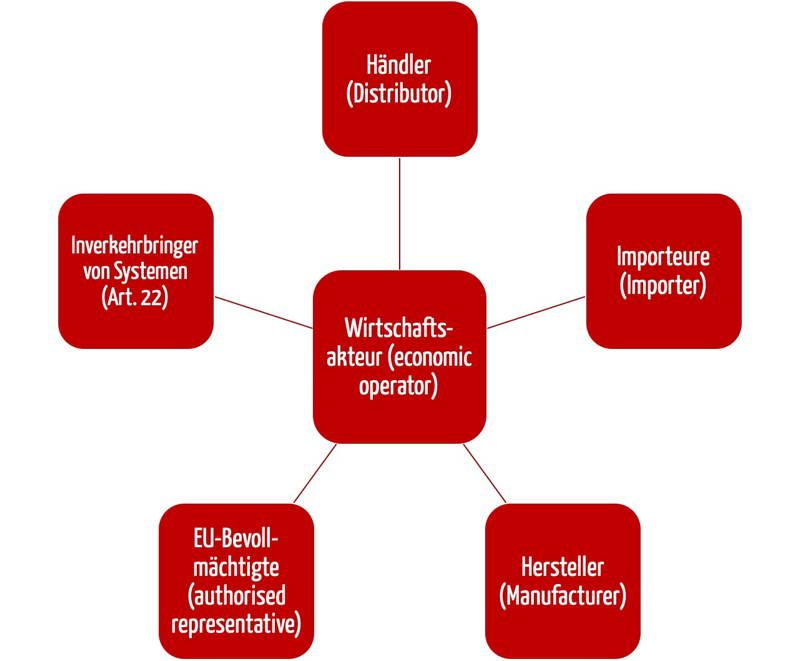
Die Medizinprodukteverordnung (MDR) hat viele Rollen eingeführt, darunter die der Wirtschaftsakteure. Zu diesen Wirtschaftsakteuren zählen neben den Herstellern, den EU-Bevollmächtigten und den Importeuren auch die Händler.
„Händler“ bezeichnet jede natürliche oder juristische Person in der Lieferkette, die ein Produkt bis zum Zeitpunkt der Inbetriebnahme auf dem Markt bereitstellt, mit Ausnahme des Herstellers oder des Importeurs
Quelle: MDR, Artikel 2
Die Händler sind somit die Personen oder Organisationen, die Medizinprodukte verkaufen, sei es an Endverbraucher oder an Zwischenhändler.
b) Typische Tätigkeiten beim Vertrieb von Medizinprodukten
Zu den typischen Tätigkeiten eines Händlers zählen:
- Kauf der Medizinprodukte vom Hersteller oder anderen (Zwischen-)Händlern
- Marketing und Verkauf von Medizinprodukten an Endkunden oder andere Händler
- Lagerung
und Transport von Produkten - Ggf. Aufbringen eigener Labels (Vorsicht damit! Dazu später mehr.)
- Einweisung
von Anwendern - Unterstützung
bei der Installation und Inbetriebnahme - Beantworten
von Fragen der Anwender - Behandeln
von Kundenbeschwerden, Rückmeldung an Hersteller
Manche Händler übernehmen auch die Wartung und Reparatur der
Produkte oder organisieren dies.
c) Abgrenzung der Rollen Händler und Importeur
Wenn ein Händler Produkte von einem Hersteller oder einem anderen Händler einkauft, der nicht in der EU angesiedelt ist, übernimmt dieser Händler zusätzlich die Rolle des Importeurs. Die MDR definiert Importeure wie folgt:
„Importeur“ bezeichnet jede in der Union niedergelassene natürliche oder juristische Person, die ein Produkt aus einem Drittland auf dem Unionsmarkt in Verkehr bringt
Quelle: MDR, Artikel 2
An Importeure stellt die MDR zusätzliche Anforderungen.
d) Abgrenzung der Rollen Händler und Hersteller
Viele Händler möchten unter eigenem Namen die Medizinprodukte verkaufen und den eigentlichen Hersteller nicht nennen. Damit wird der Händler zum Hersteller, denn die Medizinprodukteverordnung definiert Hersteller so:
„Hersteller“ bezeichnet eine natürliche oder juristische Person, die ein Produkt herstellt oder als neu aufbereitet bzw. entwickeln, herstellen oder als neu aufbereiten lässt und dieses Produkt unter ihrem eigenen Namen oder ihrer eigenen Marke vermarktet
Quelle: MDR, Artikel 2
Mit der MDR gehören auch die PLM-OEM-Konstrukte der Vergangenheit an. Dafür bietet die MDR gemäß Artikel 16, Absatz 2 den Händlern neue Möglichkeiten. Sie schätzt folgende Tätigkeiten nicht mehr als eine Produktänderung ein, die Auswirkung auf die Konformität des Produkts hat:
- Bereitstellung
von Informationen für ein Produkt. Das gilt auch für Übersetzungen. - Änderung
der äußeren Verpackung
Natürlich wäre es auch denkbar, dass der Hersteller das Produkt im „Design“ des Händlers in den Verkehr bringt. Der Hersteller muss aber immer (auch) auf dem Label erkennbar sein. Das fordert Artikel 16, Absatz 3.
2. Pflichten der Händler
Bereits bevor die Händler die Medizinprodukte verkaufen, müssen sie viele gesetzliche Anforderungen erfüllen.
a) Hintergrund und Historie
Die Anforderungen, die die Medizinprodukteverordnung MDR an
die Händler stellt, leiten sich aus einem übergeordneten Rahmenwerk zur
Vermarktung von Produkten ab. Dieses ist auch als „Goods Package“ bekannt und geht
zurück auf:
b) Pflichten der Händler als Prüfinstanz in der Lieferkette (aus Sicht der MDR)
Der Gedanke der MDR besteht darin, dass jeder Wirtschaftsakteur nach Möglichkeit sicherstellt, dass der Wirtschaftsakteur einen Schritt zuvor in der Lieferkette die regulatorischen Anforderungen erfüllt hat.

Zu diesen Überprüfungen, die die MDR von den Händlern verlangt, zählen:
- Trägt das Produkt eine CE-Kennzeichnung?
- Wurde für das Produkt eine Konformitätserklärung ausgestellt?
- Hat der Importeur seinen Namen und seine Anschrift auf dem Produkt, der Verpackung oder einem Dokument angegeben?
- Hat der Importeur mit seinen zusätzlichen(!) Kennzeichnungen nicht die Kennzeichnungen des Herstellers verdeckt?
- Hat der Hersteller die UDI vergeben?
- Scheint das Produkt konform mit den gesetzlichen Vorgaben zu sein?
Wenn eine dieser Bedingungen nicht erfüllt ist, darf der
Händler das Produkt nicht bereitstellen und muss ggf. den Hersteller, den
Importeur und den EU-Repräsentanten informieren.
Händler dürfen sich damit nicht mehr blind auf die Hersteller und Importeure verlassen. Sie sollten insbesondere in der Übergangszeit genau überprüfen, ob die Hersteller gültige Zertifikate vorlegen.
c) Pflichten der Händler als Prüfinstanz in der Lieferkette (aus Sicht des MPDG)
Anforderungen des MPDG
Die MDR erlaubt den Händlern in Artikel 14 Absatz 2, Stichprobenprüfungen durchzuführen. Sie sollen damit überprüfen können, ob die Produkte tatsächlich eine CE-Kennzeichnung, eine Konformitätserklärung und eine UDI haben.
Das Medizinproduktedurchführungsgesetz MPDG erscheint rigider: Es droht in § 92 Abs. 1 Nr. 3 in Verbindung mit § 13 MPDG mit einer Freiheitsstrafe, wenn jemand gefälschte Produkte anbietet, auf Lager hält oder in Betrieb nimmt. Das betrifft die Händler.
Das Verbot in § 13 MPDG und die dazugehörige Strafandrohung ist im Zusammenhang mit Art. 14 Abs. 2 Unterabsatz 3 MDR zu sehen: Danach bestehen ein Vertriebsstopp und eine zusätzliche Meldepflicht an die zuständige Behörde, wenn ein Händler „der Auffassung ist oder Grund zur Annahme“ hat, dass ein Produkt gefälscht ist und damit nicht konform.
Der Händler hat unserer Einschätzung nach jedoch weder hieraus noch aus § 92 Abs. 1 Nr. 3 und § 13 MPDG die regulatorische Pflicht, über die stichprobenartigen Prüfungen hinaus auf etwaige Fälschungen zu prüfen, d. h. per Default misstrauisch zu sein und darauf basierend eine 100%-Prüfung festzulegen. Für die Strafvorschrift kommt es unserer Einschätzung nach auch darauf an, ob das unzulässige Anbieten oder Lagerhalten tatsächlich gefälschter Ware fahrlässig oder gar vorsätzlich erfolgte. Es wird somit kritisch, wenn der Händler weitere Anhaltspunkte oder Verdachtsmomente aufzeigt, dass er Fälschungen vertreibt.
Sich daraus ergebende mögliche Konflikte
Wenn der Händler ein gefälschtes Produkt entdeckt und im Lager behält, macht er sich strafbar. Nach § 13 MPDG ist jedoch auch das Zurücksenden fraglich, denn dann bringt er es ggf. „aus dem Geltungsbereich dieses Gesetzes“. Das ist auch strafbar.
Dann bleibt noch die Möglichkeit, es zu vernichten. Doch auch in diesem Fall macht er sich vermeintlich strafbar, weil er den Behörden „auf Ersuchen unentgeltliche Proben des Produkts zur Verfügung stellen“ oder, „sofern dies nicht praktikabel ist, Zugang zu dem Produkt verschaffen muss“ (MDR Artikel 14(6)).
Ein Leser, der auf diesen Konflikt aufmerksam machte, schreibt:
„Da bin ich mal auf die Handreichung des Gesetzgebers gespannt, wie diese Widersprüche von einem Händler gelöst werden sollen, z.B. vom Aldi, der ja auch ein- bis zweimal im Jahr Medizinprodukte (z. B. Blutdruckmessgeräte) im Rahmen irgendwelcher Aktionen führt.“
d) Anforderungen an die Tätigkeiten der Händler
Die MDR verpflichtet die Händler zu weiteren Tätigkeiten:
- Produkte nur gemäß den Vorgaben des Herstellers lagern und transportieren
- Beschwerden sowie Berichte über Vorkommnisse sammeln und an die Hersteller und ggf. Importeure weiterleiten. Das gilt auch für Produkte, von denen die Händler selbst Zweifel an der Konformität haben.
- Ein „Register“ nichtkonformer Produkte, Rückrufe und Rücknahmen führen
- Behörden (in Deutschland BfArM gemäß § 44 MPDG) über unsichere und gefälschte Produkte sowie über Korrekturmaßnahmen informieren und diesen auf Verlangen Informationen und Unterlagen aushändigen
e) Registrierung von Händlern
Die MDR spricht in Artikel 30 zwar von dem „Elektronischen System für die Registrierung von Wirtschaftsakteuren“. Sie verpflichtet darin aber nur die Hersteller, Bevollmächtigten und Importeure. Die MDR überlässt es den Mitgliedsstaaten, Bestimmungen für die Registrierung von Händlern zu erlassen.
Genau das scheinen Mitgliedsstaaten wie Deutschland auch zu tun: Im Medizinproduktedurchführungsgesetz MPDG heißt es:
(1) Das Bundesministerium für Gesundheit wird ermächtigt, …
9. festzulegen, dass Händler, die Produkte auf dem deutschen Markt bereitstellen, dies vor Aufnahme ihrer Tätigkeit bei der zuständigen Behörde anzuzeigen haben, sowie Inhalt und Form der Anzeige zu regeln.
MPDG § 88
Damit ist eine nationale Verordnung zu erwarten, die diese
Registrierungspflicht beschreiben wird.
f) Sonderfall Qualitätsmanagementsystem
Wann Händler ein Qualitätsmanagementsystem benötigen
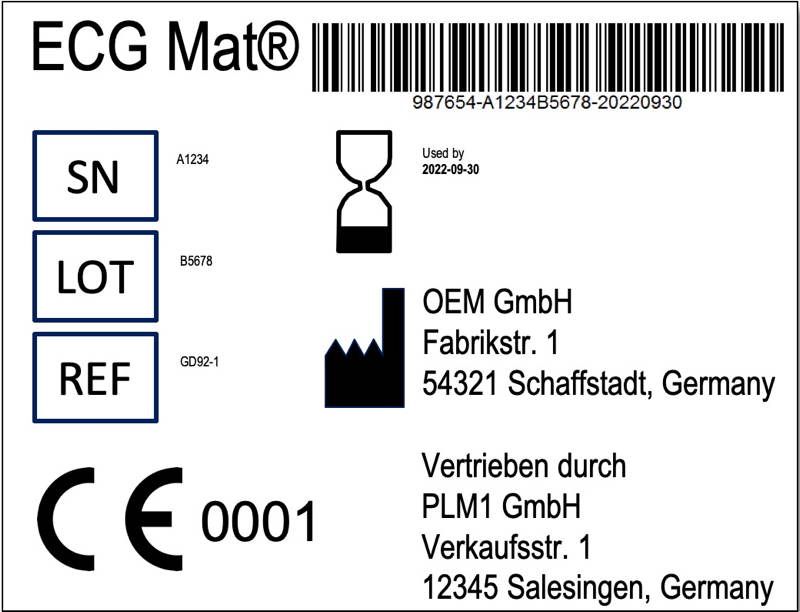
Wie oben beschrieben, dürfen Händler Produkte umverpacken und gesetzlich geforderte Informationen (Anhang I, Abschnitt 23) bereitstellen und übersetzen. Daraus ergibt sich aber nicht nur die bereits erwähnte Pflicht, den Namen des Händlers auf dem Label anzugeben (s. Abb. 2). Vielmehr muss der Händler über ein Qualitätsmanagementsystem verfügen.
Dieses QM-System muss alle relevanten Tätigkeiten regeln:
- Umverpacken des Produkts (und prüfen, dass dieses Umverpacken die Konformität nicht beeinträchtigt)
- Erstellen/Bereitstellen der Informationen (und prüfen, dass diese den gesetzlichen Anforderungen genügen)
- Übersetzen
der Informationen - Informationen über Korrekturmaßnahmen des Herstellers erhalten/einfordern(?)
Nach Aussage der irischen Behörde muss das QM-System nicht zertifiziert sein: „it is not a requirement that a quality system is officially accredited to any specific standard“. Das steht jedoch im Widerspruch zur Forderung der MDR
Within the same period of 28 days, the distributor or importer shall submit to the competent authority a certificate, issued by a notified body designated for the type of devices that are subject to activities mentioned in points (a) and (b) of paragraph 2, attesting that the quality management system of the distributor or importer complies with the requirements laid down in paragraph 3.
MDR Artikel 16, Absatz 4
Welche Prozesse ein Qualitätsmanagementsystem festlegen sollte
Ob dieses QM-System einen bestimmten Standard (z. B. einer Norm) erfüllen und nach einem bestimmten Standard geprüft werden muss, erscheint unklar. Nach Ansicht der irischen Behörde muss dieses QM-System relativ umfassend sein und auch weitere Tätigkeiten lenken, z. B.:
- Personal,
Training - Lenkung
von Dokumenten und Aufzeichnungen - Empfang,
Lagerung und Bereitstellung von Medizinprodukten - Umgang
mit zurückgegebenen Produkten - Umgang
mit gefälschten Produkten - Rückruf
von Produkten - Ausgelagerte
Prozesse - Transport
- Audits
- Lieferantenlenkung
- Management
Review - CAPA
- Abfallmanagement
- Interne
und externe Audits - Validierung inkl. Computerized Systems Validation
Beachten Sie als
- Händler: Gemäß der Behörde müssen Sie ein vollständiges QM-System vergleichbar ISO 13485 implementieren.
- Händler: Sie müssen im Fall von Artikel 16(4) der Behörde bescheinigen, dass ihr QM-System den Anforderungen ebendieses Artikels genügt.
In einer früheren Version des Artikels hieß es, dass der Hersteller die Bescheinigung vorlegen müsse. Dies ist nicht zutreffend. Es ist der Händler. Die deutsche Übersetzung ist leider falsch. Sie schreibt „Der Hersteller oder Importeur legt der zuständigen Behörde im selben Zeitraum von 28 Tagen eine Bescheinigung vor.“ Im englischen Text heißt es: „distributors or importers carrying […] shall inform the manufacturer and the competent authority of the Member State.“ Zudem wurde die Diskussion ergänzt, ob und wenn ja nach welchem Standard ein QM-System zu errichten und zu prüfen sei.
Wahrscheinlich damit die Behörde das QM-System prüfen kann, verpflichtet die MDR den Händler, sie 28 Tage vor der Bereitstellung des Produkts zu unterrichten. Die MDR spricht von „unterrichten“, nicht von „um Genehmigung bitten“.
Wir unterstützen Hersteller ebenso wie Händler dabei, schnell ISO 13485-konforme QM-Systeme zu etablieren und damit die gesetzlichen Auflagen zu erfüllen.
Kontakt aufnehmen
g) Pflichten, die die MDR den Händlern nicht(?) auferlegt
Die Händler sind nicht verpflichtet, die Produkte zu
registrieren. Das ist die Aufgabe der Hersteller bzw. Importeure. Die Händler
benötigen auch keine „Person Responsible for Regulatory Compliance“.
Unklar ist, ob der Händler für die Lagerung und den Transport verantwortlich ist, wenn der Hersteller direkt an die Kunden des Händlers liefert. Zu Diskussionen führt auch die Frage, ob der Importeur oder der Händler für den Transport vom Importeur zum Händler verantwortlich zeichnet.
h) Übergangsfristen
Händler betrifft v. a. die sogenannte Abverkaufsregelung der MDR Artikel 120, Absatz 4. Diese soll den „Zeitraum befristen, in dem AIMDD/MDD konforme Produkte, die bereits (erstmalig) in Verkehr gebracht wurden (entweder vor GB oder mittels Art. 120 Abs. 3 MDR nach GB), z. B. durch einen Händler bereitgestellt werden dürfen.“
Nach dem 27. Mai 2025 dürfen diese Produkte nicht mehr bereitgestellt/in Betrieb genommen werden (= Endtermin). Derartige Produkte, die sich an diesem Tag noch immer in der Handelskette befinden, – d. h. noch nicht dem Endanwender (z. B. Krankenhaus) als gebrauchsfertiges Produkt zur Verfügung gestellt wurden – sind nicht mehr „handelbar“.
Art. 120 Abs. 4 MDR adressiert im Wesentlichen das „Bereitstellen“ auf dem Markt von AIMDD/MDD-konformen Produkten, nachdem sie (erstmalig) in Verkehr gebracht wurden, z. B. in der Handelskette. Er regelt nicht das “(erstmalige) Inverkehrbringen” dieser Produkte durch den Hersteller.NAKI UG1
Weiter stellt der NAKI klar, dass „der Handel mit Second-Hand-Produkten nicht von der sog. „Abverkaufsregelung“ erfasst sein soll (siehe Erwägungsgrund 32). Dies bedeutet, dass sobald ein Produkt dem Endanwender (z. B. Krankenhaus) als gebrauchsfertiges Produkt zur Verfügung gestellt wurde, das weitere Bereitstellen dieses Produktes auf dem Markt nicht dem Anwendungsbereich der MDR unterfällt.“
Die MDCG stellt in MDCG 2021-25 klar, dass die Wirtschaftsakteure ihren Pflichten nach MDR auch für die Legacy-Devices (Artikel 120, gültiges MDD-Zertifikat) nachkommen müssen. Die MDCG listet für jeden Wirtschaftsakteur die entsprechenden Artikel der MDR auf, die er beachten muss. Bei den Händlern sind das:
- Artikel 14(2) letzter Abschnitt: Hier geht es um die Informationspflichten im Rahmen der Post-Market Surveillance
- Artikel 14(4): Auch hier geht es um die Informationspflichten und das Mitwirken bei den Korrekturmaßnahmen
- Artikel 14(5): Auch hier geht es um das Mitwirken im Rahmen der Post-Market Surveillance und der damit verbundenen Maßnahmen.
- Artikel 14(6): Dieser Absatz betrifft die Kooperation der Hersteller mit den Behörden.
i) Klarstellung durch den EuGH
Es gibt ein EuGH-Urteil zu Händlerpflichten (C-10/24). Der EuGH hat klargestellt, welche Prüfpflichten „Distributoren“ nach Art. 14 MDR haben – und wo die Grenzen liegen.
Der Fall: Ein Distributor vertrieb Dental-Kompressoren mit CE-Kennzeichnung als „Maschine“ – nicht als Medizinprodukt. Das BfArM sah diese jedoch als Klasse-IIa-Zubehör. Ein Wettbewerber mahnte ab.
Die drei Kernaussagen des EuGH
- Kohärenzcheck ja, Konformitätsbewertung nein: Händler müssen anhand vorliegender Dokumente prüfen, ob CE-Kennzeichnung und DoC offensichtlich zur MDR passen – aber nicht die Herstellerbewertung wiederholen.
- Klassifizierung ist Herstellersache – aber bei NB-pflichtigen Klassen muss die vierstellige NB-Nummer geprüft werden.
- Abmahnungen lösen Prüfpflicht aus: Der Händler muss den Hersteller konsultieren und kann sich auf dessen Einschätzung verlassen, solange diese nicht offensichtlich unhaltbar ist.
Kohärenzcheck ja, Konformitätsbewertung nein: Händler müssen anhand vorliegender Dokumente prüfen, ob CE-Kennzeichnung und DoC offensichtlich zur MDR passen – aber nicht die Herstellerbewertung wiederholen.
Klassifizierung ist Herstellersache – aber bei NB-pflichtigen Klassen muss die vierstellige NB-Nummer geprüft werden.
Abmahnungen lösen Prüfpflicht aus: Der Händler muss den Hersteller konsultieren und kann sich auf dessen Einschätzung verlassen, solange diese nicht offensichtlich unhaltbar ist
- Kohärenzcheck ja, Konformitätsbewertung nein: Händler müssen anhand vorliegender Dokumente prüfen, ob CE-Kennzeichnung und DoC offensichtlich zur MDR passen – aber nicht die Herstellerbewertung wiederholen.
- Klassifizierung ist Herstellersache – aber bei NB-pflichtigen Klassen muss die vierstellige NB-Nummer geprüft werden.
- Abmahnungen lösen Prüfpflicht aus: Der Händler muss den Hersteller konsultieren und kann sich auf dessen Einschätzung verlassen, solange diese nicht offensichtlich unhaltbar ist.
3. Hinweise für Hersteller (bzgl. Händler)
a) Problemstellung
Manche Hersteller mögen denken, dass all diese regulatorischen
Vorgaben das Problem der Händler und nicht das eigene seien. Doch das wäre zu
kurz gedacht:
Händler dürfen Produkte sogar „umkennzeichnen“ und „umverpacken“, z. B. im eigenen Design. Sie müssen die Hersteller nur darüber „unterrichten“, nicht um Genehmigung fragen.
Mit anderen Worten: Die Hersteller können einerseits diese Tätigkeiten nicht (einfach) unterbinden. Andererseits sind sie zur Post-Market Surveillance verpflichtet. Das wird schwierig, wenn Händler Produkte in weiteren, dem Hersteller nicht bekannten Ländern mit übersetzten Begleitmaterialien vertreiben.
b) Post-Market Surveillance
Hersteller sollten proaktiv und weltweit nach Informationen zu den eigenen Produkten suchen. Das betrifft explizit nicht die Länder, von denen sie wissen, dass ihre Produkte vertrieben werden. Hier hilft eine Automatisierung wie das Post-Market Radar des Johner Instituts.
Es ist unerlässlich, dass die Hersteller mit ihren Händlern,
Bevollmächtigten und Importeuren genau regeln, wer für welche Tätigkeiten (nicht
nur) im Rahmen der Post-Market Surveillance zuständig ist.
Dass diese Überwachung nach der Inverkehrbringung auch
gefälschte Produkte und nicht autorisierte Händler im Fokus haben sollte,
versteht sich von selbst.
c) Händler an die Kandare nehmen
Hersteller sollten die Händler genauso überwachen wie die
Produkte selbst.
Händlern, die die Produkte „umverpacken“ oder „umkennzeichnen“, müssen der Behörde sogar ein Zertifikat einer Benannten Stelle vorlegen. Damit ist den Herstellern ein Teil der Arbeit abgenommen.
In einer früheren Version des Artikels hieß es, dass die Hersteller die Zertifikate vorlegen müssten. Dies ist allerdings ein Fehler in der deutschen Version der MDR. Dort hieß es „Der Hersteller oder Importeur legt…“, in der englischen Version heißt es „the distributor or importer shall submit …“
Dennoch können sich die Hersteller nicht blind darauf verlassen. Sie müssen ihre Händler kennen und überwachen.
Manchmal ist es hilfreich, den bestimmungsgemäßen Gebrauch und das Design der Produkte (z. B. Stecker) so zu wählen, dass bestimmte Märkte ausgeschlossen sind. Damit können Hersteller den „Missbrauch“ durch ungewünschte Händler etwas eindämmen.
Zudem sollten die Hersteller mit den Händlern Qualitätssicherungsvereinbarungen abschließen. So eine QSV sollte z. B. regeln:
- Betroffene
Produkte bzw. Produktgruppen - Geltungsdauer
der Vereinbarung - Ansprechpartner
auf beiden Seiten - Form
und Fristen für Rückmeldungen der Händler an den Hersteller - Recht des Herstellers auf Audit/Inspektion des Händlers
- Verpflichtung
des Händlers zu Behördenmeldungen - Verpflichtung
des Händlers zum Nachverfolgen der Produkte („Register“) - Ggf.
Verbot zum Verkauf an andere Händler - Verpflichtung
des Händlers, Importeure und EU-Beauftrage zu informieren - Bereithalten
von Prüfmustern - Verpflichtung,
Begleitmaterialien bereitzulegen - Verpflichtung,
nur mit einer zertifizierten Übersetzungsagentur zu arbeiten
d) Vorsicht mit Fulfillment-Partnern
Bestimmte Logistikpartner bieten Dienste an, die über die eines üblichen Paketdienstleisters (z. B. DHL) hinausgehen. Diese sogenannten „Fulfillment Centers“ oder „Fulfillment Houses“ lagern Produkte, verpacken diese auf Wunsch, übernehmen die Fakturierung usw.
Dann spricht man nicht mehr von „neutralen Dienstleistern“,
sondern von Händlern im Sinne der MDR.
Die MDR hat die Anforderungen an die Händler substanziell erhöht. Das ist nachvollziehbar, weil nur alle an der Logistikkette Beteiligten gemeinsam nachvollziehen können, wo sich welche Produkte befinden und ob diese Produkte konform sind.
4. FAQ
a) Wer darf Medizinprodukte verkaufen?
Der Gesetzgeber schließt keine Personen oder Organisationen vom Verkauf von Medizinprodukten aus. Allerdings stellt der Gesetzgeber Anforderungen an diese Verkäufer und nennt diese Händler. Die Händlerpflichten wurden in diesem Artikel vorgestellt.
Medizinproduktehersteller können jedoch den Handel mit ihren Produkten (mit Einschränkungen) untersagen.
b) Welche Erlaubnis oder Berechtigung ist erforderlich, um mit Medizinprodukten handeln zu dürfen?
Es gibt keine offizielle Erlaubnis oder Berechtigung durch staatliche Stellen. Allerdings müssen die Händler die gesetzlichen Voraussetzungen erfüllen, bevor sie mit dem Handel beginnen dürfen. Zu diesen Voraussetzungen können zählen:
- Qualitätsmanagementsystem (für bestimmte Händler)
- Registrierung bei Behörden (z. B. in Deutschland; siehe oben)
- Prüfungen des Produkts (z. B. Angaben des Importeurs, UDI)
Falls ein Händler auch Importeur ist, kommen weitere Voraussetzungen hinzu, die er vor der Aufnahme des Handels erfüllen muss.
Zudem kann es sein, dass ein Hersteller Anforderungen an die Händler stellt. Es könnte etwa sein, dass der Händler einen Kompetenznachweis vorlegt oder beim Hersteller erbringt.
c) Wie bringt man ein Medizinprodukt auf den Markt?
Die EU unterscheidet genau zwischen der Inverkehrbringung, der erstmaligen Inverkehrbringung und der Bereitstellung. Händler sind die natürlichen oder juristischen Personen, die Produkte auf dem Markt bereitstellen.
5. Fazit / Zusammenfassung
Nur gemeinsam können die Wirtschaftsakteure auf Nichtkonformitäten reagieren und so die Sicherheit der Patientinnen und Patienten erhöhen. Damit nimmt ihnen die MDR die Möglichkeiten eines „Finger Pointings“.
Das sind wichtige Erkenntnisse:
- Die Händler sollten über ein (weitgehend) vollständiges Qualitätsmanagementsystem verfügen. Das ist zumindest die Sicht der irischen Behörde. Nicht nur für das Rückmeldesystem sollten Prozesse etabliert sein.
Für den Fall, dass ein Händler Tätigkeiten gemäß Artikel 16(2) durchführt, muss er über ein QM-System verfügen. Eine Pflicht zur Zertifizierung gibt es (derzeit) nicht. - Die Hersteller sollten sich bewusst sein, dass die Pflichten der Händler sie zumindest indirekt ebenfalls betreffen.
- Die irische Behörde hat eine Leitlinie für Händler mit weiteren Praxistipps veröffentlicht.
So bleibt zu hoffen, dass das MPDG nicht zu Widersprüchen mit der MDR führt und dass eine höhere Sicherheit der Patienten und nicht ein Mehr an Bürokratie das Ergebnis dieser gesetzlichen Verschärfungen sind.
- 2026-06-11: Abschnitt 2.i) eingefügt, der das neue EuGH-Urteil berücksichtigt
- 2024-12-18: FAQ (Kapitel 4) eingefügt. Zwischenüberschriften bei 2.e) und 2.f) eingefügt. Hauptüberschriften gekürzt. Einige redaktionelle Verbesserungen.
- 2021-10-25: In Abschnitt 2.h) die Anforderungen durch die Leitlinie MDCG 2021-25 ergänzt
Ähnliche Beiträge
PakarPBN
A Private Blog Network (PBN) is a collection of websites that are controlled by a single individual or organization and used primarily to build backlinks to a “money site” in order to influence its ranking in search engines such as Google. The core idea behind a PBN is based on the importance of backlinks in Google’s ranking algorithm. Since Google views backlinks as signals of authority and trust, some website owners attempt to artificially create these signals through a controlled network of sites.
In a typical PBN setup, the owner acquires expired or aged domains that already have existing authority, backlinks, and history. These domains are rebuilt with new content and hosted separately, often using different IP addresses, hosting providers, themes, and ownership details to make them appear unrelated. Within the content published on these sites, links are strategically placed that point to the main website the owner wants to rank higher. By doing this, the owner attempts to pass link equity (also known as “link juice”) from the PBN sites to the target website.
The purpose of a PBN is to give the impression that the target website is naturally earning links from multiple independent sources. If done effectively, this can temporarily improve keyword rankings, increase organic visibility, and drive more traffic from search results.